Modeling of dynamically rearranging networks made easy
Supercol ESR Rodrigo Rivas Barbosa has improved the modeling of dynamically rearranging molecular structures by means of bond-swapping. He created a plugin for the open-source molecular dynamics software LAMMPS that is freely available on GitHub. This achievement marks milestone 4 of SuperCol: “Software to model particle-particle interactions”.
The plugin greatly enhances the simulation of molecular rearrangements that take place for instance in vitrimers and other topologically adaptable networks. Such novel materials combine two essential features: on the one hand they are strong due to (covalent) bonds between molecules and particles, on the other hand they are self-healing due to swapping of these bonds.
Theoretical and numerical studies of the dynamics of such adaptable materials require effective algorithms for modeling the corresponding evolution of bonds. Rodrigo Rivas Barbosa (ESR 15) has now developed a molecular dynamics simulation of such a bond-swapping network with particle-level description. His LAMMPS plugin represents the bond-swapping mechanism that underlies the dynamical rearragements by means of implementating two different three-body potentials. This facilitates the modeling process and substantially speeds up the simulation. The three-body potentials are based on earlier work[1] by our SuperCol PI Francesco Sciortino at the Physics Department of Sapienza Università di Roma where Rivas Barbosa is currently doing his PhD research.
Available on GitHub
The method is computationally cheap, implicitly accounts for the single-bond per particle condition, can keep the potential energy unchanged throughout the bond-swapping process, and can even tune the bond-swapping event frequency. Taking advantage of LAMMPS parallel computing, the running time of simulations can be further reduced. The plugin is hosted in Github and is freely available for download. The potential parameters are flexible and easy to set, and a guide and an example folder are provided to help familiarize the users with the new exciting features.
[1] Sciortino, F. Three-body potential for simulating bond swaps in molecular dynamics. Eur. Phys. J. E 40, 3 (2017). DOI: 10.1140/epje/i2017-11496-5

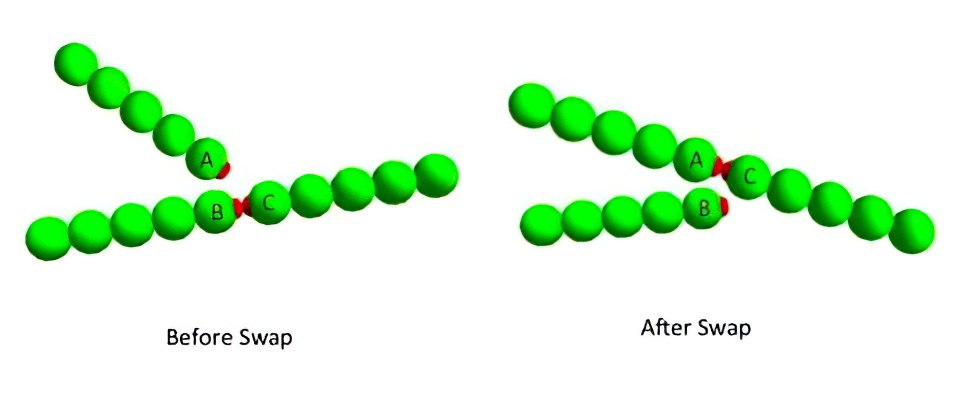
Example of a bond-swapping event via the three-body potential:
- Initially particle 1 and 2 are bonded since they are separated by a distance smaller than the cutoff radius (orange circle); particle 3 is outside the interaction range.
- Then, particle 3 gets inside particle 1 cutoff radius, the three-body potential is activated, and the possibility of a bond-swapping event is in line.
- Finally, the bond is rearranged to hold together particles 3 and 1, particle 2 leaves the interaction range and the three-body potential is turned back off.
Related posts


